Training-3
Scalable Approaches for Large-Scale Genetic Association Analyses
Contents
- Brief Overview of GWAS
- Installing the Software and Dataset
- Introduction to fastGWA and fastGWA-GLMM
- Running GWAS with fastGWA and fastGWA-GLMM
- ☕ Coffee break (15 mins)
- Introduction to REGENIE
- Running GWAS with REGENIE
- Introduction to CMA
- Running GWAS with CMA
- (Optional) Running Meta-Analysis with CMA
- 💬 Questions / Comments / Feedback
- References
⬤ Coding
Session
⬤
Concepts & Theory
- Brief Overview of GWAS
A genome-wide association study (GWAS) is a research approach used to identify genetic variants associated with specific traits or diseases by scanning the genomes of many individuals. In GWAS, researchers compare the frequency of single-nucleotide polymorphisms (SNPs) across the entire genome between individuals with different trait values, such as cases versus controls for a disease (binary traits) or variations in a continuous measurement like height or blood pressure (quantitative traits). This method can reveal genetic risk factors without prior knowledge of candidate genes, making it especially powerful for studying complex traits influenced by many genes and environmental factors. Insights from GWAS contribute to understanding biological pathways, improving disease risk prediction, and guiding precision medicine efforts.
Simple Linear Regression in GWAS:
|
|
\[ y = g \cdot \beta_g + \epsilon \] Where:
|
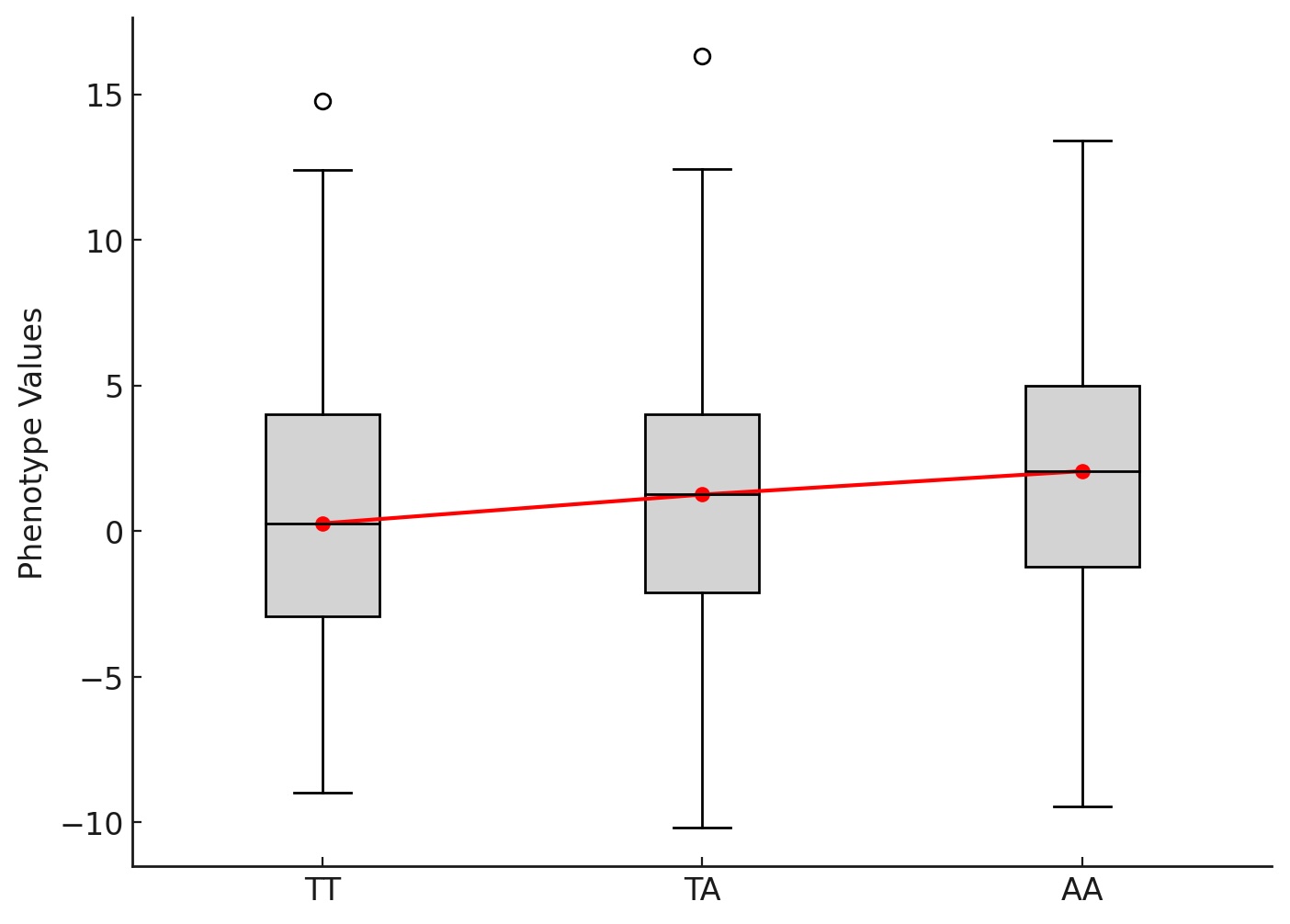
Confounders in GWAS:
- Population structure
- Related individuals
- Linkage disequilibrium (LD)
Check the YouTube video for more information: Watch on YouTube
Linear Mixed Models (LMMs) in GWAS
\[ y = X\beta + g_j \beta_j + u + \epsilon \]
Where:
- \(y\): Phenotype vector (\(n \times 1\)) for all individuals
- \(X\beta\): Fixed effects for
non-genetic covariates (e.g., age, sex, principal components)
- \(g_j \beta_j\): Fixed effect of
the \(j\)-th genetic variant being
tested
- \(u\): Polygenic random effect,
\(u \sim N(0, \sigma_g^2 K)\)
- \(K\): Genetic relationship matrix (GRM), often stored in sparse form for efficiency (it can be computed from genotype data)
- \(\sigma_g^2\): Genetic variance
component
- \(\epsilon\): Residual error, \(\epsilon \sim N(0, \sigma_e^2 \mathbf{I})\)
Solving LMMs in GWAS
The generalised least squares estimate \(\widehat{\beta}_j\) of the effect size of \(g_j\) is obtained as:
\[ \widehat{\beta}_j = \frac{g_j^{T} \mathbf{V}^{-1} y}{g_j^{T} \mathbf{V}^{-1} g_j} \]
where \(\mathbf{V}\) is the
variance–covariance matrix of the phenotype, incorporating both
polygenic and residual variance components:
\[
\mathbf{V} = K\cdot\sigma_g^2+\mathbf{I}\cdot\sigma_e^2
\]
The corresponding variance of \(\widehat{\beta}_j\) is:
\[ \mathrm{Var}(\widehat{\beta}_j) = (g_j^{T} \mathbf{V}^{-1} g_j)^{-1} \] The only unknowns in this solution are the variance components \(\sigma^2_g\) and \(\sigma^2_e\), which are estimated using efficient restricted maximum likelihood (REML) methods.
Hypothesis Testing in GWAS (LMMs)
In GWAS, statistical hypothesis testing is performed to determine
whether a given genetic variant is associated with a trait of
interest.
For each variant, we assess the following competing hypotheses:
\[ \begin{aligned} H_0: & \quad \beta_j = 0 & \text{(no association between variant \( g_j \) and phenotype \( y \))} \\ H_1: & \quad \beta_j \neq 0 & \text{(variant \( g_j \) is associated with \( y \))} \end{aligned} \]
Under the null hypothesis \(H_0\), the squared Wald statistic is given by: \[ T_j^2 = \frac{\widehat{\beta}_j^2}{\mathrm{Var}(\widehat{\beta}_j)} \;\sim\; \chi^2_1 \] where \(\widehat{\beta}_j\) is the estimated effect size and \(\mathrm{Var}(\widehat{\beta}_j)\) its variance.
The statistical significance for variant \(j\) is assessed by: \[ p_j = 1 - F_{\chi^2_1}(T_j^2) \] where \(F_{\chi^2_1}(\cdot)\) is the cumulative distribution function of the chi-square distribution with 1 degree of freedom.
Evidence of Association in GWAS LMMs
A small \(p\)-value suggests rejecting \(H_0\) in favour of \(H_1\), meaning the variant is more likely to be associated with the trait. This naturally leads to the question: how small must the \(p\)-value be to consider the evidence convincing?
In GWAS, where millions of variants are tested, a stringent genome wide significance threshold is applied, often \(p < 5 \times 10^{-8}\). This threshold approximates a Bonferroni correction for about one million effectively independent tests and addresses the challenge known as the multiple testing problem in GWAS.
Why use a stringent threshold in GWAS?
In GWAS, millions of variants (\(M\)) are tested across the genome. The expected number of false positives is approximately \(M \times \alpha\), where \(\alpha\) is the chosen significance level.
Without adjustment, using a conventional \(\alpha = 0.05\) would yield thousands of false positives purely by chance.
This motivates the adoption of a much smaller threshold, such as \(5 \times 10^{-8}\), to keep the average number of false positives at a reasonable level.
Key idea in LMMs:
Linear mixed models account for both fixed and random effects, helping
to control for confounders while efficiently testing the association
between each genetic variant and the phenotype.
Note on LOCO (Leave-One-Chromosome-Out):
GWAS methods use a LOCO scheme when generating polygenic predictions or fitting random effects.
In LOCO, the genetic relationship matrix (or polygenic score) is calculated excluding the chromosome currently being tested.
This prevents “proximal contamination” [6].
GWAS Requires Large Sample Sizes
Genome-wide association studies typically test millions of genetic variants across the genome. Because each variant often explains only a very small fraction of the variation in a trait, detecting true associations with sufficient statistical power requires large numbers of participants. Larger sample sizes reduce random noise, improve the precision of effect size estimates, and help identify variants with small effects that would be missed in smaller datasets. This scale of data creates a strong need for highly efficient and scalable computational tools that can process and analyse results within a reasonable time frame and with optimal computational resources.
- Installing the Software and Dataset
This training course covers scalable GWAS tools REGENIE, fastGWA/fastGWA-GLMM, and CMA, all using the same prepared dataset and a shared set of software dependencies. Follow the steps below to set up your environment, install the required software, and download the dataset for use throughout the course.
-
Clone the CMA Repository (includes setup instructions for the
software and dataset)
git clone https://git.ecdf.ed.ac.uk/cma/snake-cma.git -
Install dependencies — choose one of the following:
-
Using Conda (recommended)
conda env create -f snake-cma/environment.yml conda activate snake-cma -
Using Pip
pip install -r snake-cma/requirements.txt
-
Using Conda (recommended)
-
Download the example dataset and software binaries
The provided scripts will download static versions of PLINK, REGENIE, and GCTA (contains fastGWA and fastGWA-GLMM), and generate the HapMap3-based dataset that will be used in all hands-on sessions.
This will create:chmod +x ./snake-cma/examples/download_software.sh ./snake-cma/examples/generate_dataset.sh bash ./snake-cma/examples/download_software.sh bash ./snake-cma/examples/generate_dataset.sh-
./snake-cma/examples/software/— Contains required software: REGENIE, fastGWA/fastGWA-GLMM, and CMA -
./snake-cma/examples/data/— Contains required dataset used in exercises
-
Before starting any tutorial, ensure your snake-cma
environment is active (if not already):
conda activate snake-cma
Next, save the current working directory (location of the cloned
snake-cma repository) into a variable. This will be reused
throughout the tutorial.
dir_user=$(pwd)At this point, you can quickly check that the example dataset has been generated correctly by listing its contents:
ls -lh ${dir_user}/snake-cma/examples/data
The data folder should contain the following files:
| File | Content |
|---|---|
binary_trait.phen |
Phenotype file for a binary trait (BT). |
continuous_trait.phen |
Phenotype file for a quantitative trait (QT). |
HAPMAP3.covars.cov |
Covariate file for REGENIE. |
HAPMAP3.covars.qcovar |
Covariate file for fastGWA/fastGWA-GLMM (Quantitative) |
HAPMAP3.covars.bcovar |
Covariate file for fastGWA/fastGWA-GLMM (Discrete) |
HAPMAP3.qc.genotype.{bed,bim,fam} |
Genotype files in PLINK binary
format (.bed, .bim,
.fam). |
- Introduction to fastGWA and fastGWA-GLMM
fastGWA is an efficient mixed linear model (MLM) approach implemented in the GCTA software for genome-wide association studies that can handle large cohorts while accounting for population structure and relatedness. It achieves speed by using a sparse genetic relationship matrix (GRM), which stores only relationships above a certain relatedness threshold, dramatically reducing memory and computational costs compared to a full GRM. This makes fastGWA well suited for quantitative traits in biobank-scale datasets.
fastGWA-GLMM is an extension of fastGWA designed for binary traits, using a generalized linear mixed model to correctly model case–control outcomes while still leveraging the sparse GRM for computational efficiency. By fitting the model on the observed binary phenotype and using approximate inference, fastGWA-GLMM can scale to millions of variants and hundreds of thousands of individuals while maintaining control over confounding from relatedness and population stratification.Linear Mixed Model for Quantitative Traits
For a given genetic variant \(j\):
\[ y = X\beta + g_j \alpha_j + u + \epsilon \]
Where:
- \(y\): Phenotype vector (\(n \times 1\))
- \(X\): Covariate matrix (e.g., age,
sex, principal components)
- \(\beta\): Fixed effect
coefficients for covariates
- \(g_j\): Genotype vector for
variant \(j\) (0, 1, 2 dosage)
- \(\alpha_j\): Effect size of
variant \(j\)
- \(u\): Polygenic random effect,
\(u \sim N(0, \sigma_g^2 K)\)
- \(K\): Genetic relationship matrix
(GRM), often sparse for efficiency
- \(K\): Genetic relationship matrix
(GRM), often sparse for efficiency
- \(\epsilon\): Residual error, \(\epsilon \sim N(0, \sigma_e^2 \mathbf{I})\)
The null hypothesis \(H_0: \alpha_j = 0\) is tested for each variant.
Generalized Linear Mixed Model for Binary Traits
For binary phenotypes (\(y_i \in \{0,1\}\)), a logistic link is used:
\[ \text{logit}\left[ P(y = 1) \right] = X \beta + g_{j} \alpha_j + u \]
Where:
- \(\text{logit}(p) = \log \left(
\frac{p}{1-p} \right)\)
- \(u \sim N(0, \sigma_g^2 K)\)
models genome-wide background effects
- The residual variance is absorbed in the logistic model framework
Computational Note
- Both fastGWA and fastGWA-GLMM models estimate \(\sigma_g^2\) and \(\sigma_e^2\) once under
the null and reuse them across variants.
- Using a sparse GRM drastically reduces memory and
computation.
- Running GWAS with fastGWA and fastGWA-GLMM
We will follow the instructions from the GCTA website to perform GWAS using fastGWA and fastGWA-GLMM.
EXERCISE 1: Running fastGWA Interactively (Quantitative Trait)
dir_data=${dir_user}/snake-cma/examples/data
dir_cma=${dir_user}/snake-cma/examples/cma-tests
mkdir -p ${dir_cma}
geno=${dir_data}/HAPMAP3.qc.genotype
pheno=${dir_data}/continuous_trait.phen
covars=${dir_data}/HAPMAP3.covars.cov
output=${dir_cma}/ex1-gcta-qt
grm=${dir_data}/HAPMAP3.qc.genotype.grm_matrix
gcta=${dir_user}/snake-cma/examples/software/gcta64/gcta-1.94.4-linux-kernel-3-x86_64/gcta64
echo Starting..
# Build sparse GRM only if it doesn't exist
if [ ! -f "${grm}.grm.sp" ]; then
${gcta} --bfile ${geno} \
--make-grm \
--sparse-cutoff 0.05 \
--out ${grm}
fi
${gcta} --bfile ${geno} \
--grm-sparse ${grm} \
--fastGWA-mlm \
--pheno ${pheno} \
--qcovar ${covars} \
--out ${output}
echo Done..
Once the analysis is complete, you can inspect the output summary
statistics generated by fastGWA. The results file contains genome
information such as CHR (chromosome), SNP
(variant identifier), and POS (base-pair position), along
with association results such as BETA (effect size
estimate), SE (standard error of the effect size), and
P (p-value for association). These columns allow you to
interpret the strength and direction of association between each variant
and the phenotype.
EXERCISE 2: Perform GWAS with fastGWA interactively using both –qcovar and –covar options
Goal: Demonstrate how fastGWA automatically transforms discrete covariates into dummy variables and merges quantitative and discrete covariates into a single combined covariate file.
Hint
Use the–qcovar option for continuous (quantitative)
covariates and the –covar option for discrete covariates in
fastGWA. You need to use HAPMAP3.covars.qcovar and
HAPMAP3.covars.bcovar respectively.
Show solution
dir_data=${dir_user}/snake-cma/examples/data
dir_cma=${dir_user}/snake-cma/examples/cma-tests
mkdir -p ${dir_cma}
geno=${dir_data}/HAPMAP3.qc.genotype
pheno=${dir_data}/continuous_trait.phen
qcovars=${dir_data}/HAPMAP3.covars.qcovar
bcovars=${dir_data}/HAPMAP3.covars.bcovar
output=${dir_cma}/ex2-gcta-qt
grm=${dir_data}/HAPMAP3.qc.genotype.grm_matrix
gcta=${dir_user}/snake-cma/examples/software/gcta64/gcta-1.94.4-linux-kernel-3-x86_64/gcta64
echo Starting..
# Build sparse GRM only if it doesn't exist
if [ ! -f "${grm}.grm.sp" ]; then
${gcta} --bfile ${geno} \
--make-grm \
--sparse-cutoff 0.05 \
--out ${grm}
fi
${gcta} --bfile ${geno} \
--grm-sparse ${grm} \
--fastGWA-mlm \
--pheno ${pheno} \
--qcovar ${qcovars} \
--covar ${bcovars} \
--out ${output}
echo Done..EXERCISE 3: Run GWAS with fastGWA via a Bash script
Goal: Write and
run a small Bash script that builds a sparse GRM (if missing) and runs
fastGWA for a quantitative trait. Make sure to change the
output variable to a unique name before running.
Show solution
Create the script file:
nano ex3.shPaste the solution below, then save and close:
#!/bin/bash set -euo pipefail dir_user="${1:?ERROR: provide repo root as first argument}" dir_data="${dir_user}/snake-cma/examples/data" dir_cma="${dir_user}/snake-cma/examples/cma-tests" mkdir -p "${dir_cma}" geno="${dir_data}/HAPMAP3.qc.genotype" pheno="${dir_data}/continuous_trait.phen" covars="${dir_data}/HAPMAP3.covars.cov" output="${dir_cma}/ex3-gcta-qt" grm="${dir_data}/HAPMAP3.qc.genotype.grm_matrix" gcta="${dir_user}/snake-cma/examples/software/gcta64/gcta-1.94.4-linux-kernel-3-x86_64/gcta64" echo "Starting.." # Build sparse GRM only if it doesn't exist if [ ! -f "${grm}.grm.sp" ]; then "${gcta}" \ --bfile "${geno}" \ --make-grm \ --sparse-cutoff 0.05 \ --out "${grm}" fi "${gcta}" \ --bfile "${geno}" \ --grm-sparse "${grm}" \ --fastGWA-mlm \ --pheno "${pheno}" \ --qcovar "${covars}" \ --out "${output}" echo "Done.."Run the script (pass the repository root you saved earlier as dir_user):
bash ex3.sh "${dir_user}"
EXERCISE 4 (Optional): Run GWAS with fastGWA using an HPC job script on your cluster
Goal: Write and
run a small job script for your local HPC cluster that builds a sparse
GRM (if missing) and runs fastGWA for a quantitative trait. Make sure to
change the output variable to a unique name before running.
Show solution
A solution for Eddie (the University of Edinburgh HPC cluster) will be provided during the in-person training.EXERCISE 5: Running fastGWA-GLMM Interactively (Binary Trait)
dir_data=${dir_user}/snake-cma/examples/data
dir_cma=${dir_user}/snake-cma/examples/cma-tests
mkdir -p ${dir_cma}
geno=${dir_data}/HAPMAP3.qc.genotype
pheno=${dir_data}/binary_trait.phen
covars=${dir_data}/HAPMAP3.covars.cov
output=${dir_cma}/ex5-gcta-bt
grm=${dir_data}/HAPMAP3.qc.genotype.grm_matrix
gcta=${dir_user}/snake-cma/examples/software/gcta64/gcta-1.94.4-linux-kernel-3-x86_64/gcta64
echo Starting..
# Build sparse GRM only if it doesn't exist
if [ ! -f "${grm}.grm.sp" ]; then
${gcta} --bfile ${geno} \
--make-grm \
--sparse-cutoff 0.05 \
--out ${grm}
fi
${gcta} --bfile ${geno} \
--grm-sparse ${grm} \
--fastGWA-mlm-binary \
--pheno ${pheno} \
--qcovar ${covars} \
--joint-covar \
--out ${output}
echo Done..EXERCISE 6: Run GWAS with fastGWA-GLMM via a Bash script
Goal: Write and run a small Bash script that builds a sparse GRM (if missing) and runs fastGWA-GLMM for a binary trait. Make sure to change the output variable to a unique name before running.
Show solution
Create the script file:
nano ex6.shPaste the solution below, then save and close:
#!/bin/bash set -euo pipefail dir_user="${1:?ERROR: provide repo root as first argument}" dir_data="${dir_user}/snake-cma/examples/data" dir_cma="${dir_user}/snake-cma/examples/cma-tests" mkdir -p "${dir_cma}" geno="${dir_data}/HAPMAP3.qc.genotype" pheno="${dir_data}/binary_trait.phen" covars="${dir_data}/HAPMAP3.covars.cov" output="${dir_cma}/ex6-gcta-bt" grm="${dir_data}/HAPMAP3.qc.genotype.grm_matrix" gcta="${dir_user}/snake-cma/examples/software/gcta64/gcta-1.94.4-linux-kernel-3-x86_64/gcta64" echo "Starting.." # Build sparse GRM only if it doesn't exist if [ ! -f "${grm}.grm.sp" ]; then "${gcta}" \ --bfile "${geno}" \ --make-grm \ --sparse-cutoff 0.05 \ --out "${grm}" fi "${gcta}" \ --bfile "${geno}" \ --grm-sparse "${grm}" \ --fastGWA-mlm-binary \ --joint-covar \ --pheno "${pheno}" \ --qcovar "${covars}" \ --out "${output}" echo "Done.."Run the script (pass the repository root you saved earlier as dir_user):
bash ex6.sh "${dir_user}"
EXERCISE 7: Run GWAS with fastGWA-GLMM via a Bash script using only quantitative covariates
Goal: Assess how results differ when including only quantitative covariates compared to using the full set of covariates.
Show solution
Create the script file:
nano ex7.shPaste the solution below, then save and close:
#!/bin/bash set -euo pipefail dir_user="${1:?ERROR: provide repo root as first argument}" dir_data="${dir_user}/snake-cma/examples/data" dir_cma="${dir_user}/snake-cma/examples/cma-tests" mkdir -p "${dir_cma}" geno="${dir_data}/HAPMAP3.qc.genotype" pheno="${dir_data}/binary_trait.phen" covars="${dir_data}/HAPMAP3.covars.qcovar" output="${dir_cma}/ex7-gcta-bt" grm="${dir_data}/HAPMAP3.qc.genotype.grm_matrix" gcta="${dir_user}/snake-cma/examples/software/gcta64/gcta-1.94.4-linux-kernel-3-x86_64/gcta64" echo "Starting.." # Build sparse GRM only if it doesn't exist if [ ! -f "${grm}.grm.sp" ]; then "${gcta}" \ --bfile "${geno}" \ --make-grm \ --sparse-cutoff 0.05 \ --out "${grm}" fi "${gcta}" \ --bfile "${geno}" \ --grm-sparse "${grm}" \ --fastGWA-mlm-binary \ --joint-covar \ --pheno "${pheno}" \ --qcovar "${covars}" \ --out "${output}" echo "Done.."Run the script (pass the repository root you saved earlier as dir_user):
bash ex7.sh "${dir_user}"
EXERCISE 8 (Optional): Run GWAS with fastGWA using an HPC job script on your cluster
Goal: write and
run a small job script for your local HPC cluster that builds a sparse
GRM (if missing) and runs fastGWA-GLMM for a quantitative trait. Make
sure to change the output variable to a unique name before
running.
Show solution
A solution for Eddie (the University of Edinburgh HPC cluster) will be provided during the in-person training.EXERCISE 9: Run GWAS with Multiple Phenotypes using fastGWA or fastGWA-GLMM
Goal: fastGWA and
fastGWA-GLMM do not support analysing multiple phenotypes in a single
run. This exercise shows how to run one phenotype at a time using the
–mpheno option.
The example below demonstrates how to run GWAS for the second trait (Y2) when the phenotype file contains two traits, Y1 and Y2.
dir_data="${dir_user}/snake-cma/examples/data"
dir_cma="${dir_user}/snake-cma/examples/cma-tests"
mkdir -p "${dir_cma}"
geno="${dir_data}/HAPMAP3.qc.genotype"
pheno="${dir_data}/binary_trait.phen"
covars="${dir_data}/HAPMAP3.covars.qcovar"
output="${dir_cma}/ex9a-gcta-bt"
grm="${dir_data}/HAPMAP3.qc.genotype.grm_matrix"
gcta="${dir_user}/snake-cma/examples/software/gcta64/gcta-1.94.4-linux-kernel-3-x86_64/gcta64"
# Create another Phenotype file with two trait values (Y1 and Y2)
# We will be using only Y2!
pheno_v2="${dir_data}/binary_trait.v2.phen"
awk 'BEGIN{OFS=" "} NR==1{print $0,"Y2"; next} {L[NR]=$0; Y[++n]=$3} \
END{srand(); for(i=1;i<=n;i++){j=int(1+rand()*n); t=Y[i]; Y[i]=Y[j]; Y[j]=t} \
for(i=2;i<=n+1;i++){split(L[i],a,/ +/); print a[1],a[2],a[3],Y[i-1]}}' "${pheno}" > "${pheno_v2}"
echo "Starting.."
# Build sparse GRM only if it doesn't exist
if [ ! -f "${grm}.grm.sp" ]; then
"${gcta}" \
--bfile "${geno}" \
--make-grm \
--sparse-cutoff 0.05 \
--out "${grm}"
fi
"${gcta}" \
--bfile "${geno}" \
--grm-sparse "${grm}" \
--fastGWA-mlm-binary \
--joint-covar \
--pheno "${pheno_v2}" \
--mpheno 2 \
--qcovar "${covars}" \
--out "${output}"
echo "Done.."You can also loop over the two traits and run fastGWA or fastGWA-GLMM in a single script, executing two GWAS analyses back to back.
dir_data="${dir_user}/snake-cma/examples/data"
dir_cma="${dir_user}/snake-cma/examples/cma-tests"
mkdir -p "${dir_cma}"
geno="${dir_data}/HAPMAP3.qc.genotype"
pheno="${dir_data}/binary_trait.phen"
covars="${dir_data}/HAPMAP3.covars.qcovar"
output="${dir_cma}/ex9b-gcta-bt"
grm="${dir_data}/HAPMAP3.qc.genotype.grm_matrix"
gcta="${dir_user}/snake-cma/examples/software/gcta64/gcta-1.94.4-linux-kernel-3-x86_64/gcta64"
pheno_v3="${dir_data}/binary_trait.v3.phen"
awk 'BEGIN{OFS=" "} NR==1{print $0,"Y2"; next} {L[NR]=$0; Y[++n]=$3} \
END{srand(); for(i=1;i<=n;i++){j=int(1+rand()*n); t=Y[i]; Y[i]=Y[j]; Y[j]=t} \
for(i=2;i<=n+1;i++){split(L[i],a,/ +/); print a[1],a[2],a[3],Y[i-1]}}' "${pheno}" > "${pheno_v3}"
echo "Starting.."
# Build sparse GRM only if it doesn't exist
if [ ! -f "${grm}.grm.sp" ]; then
"${gcta}" \
--bfile "${geno}" \
--make-grm \
--sparse-cutoff 0.05 \
--out "${grm}"
fi
for m in 1 2; do
out=${output}".trait_${m}"
echo "Running GWAS for phenotype column ${m}..."
"${gcta}" \
--bfile "${geno}" \
--grm-sparse "${grm}" \
--fastGWA-mlm-binary \
--joint-covar \
--pheno "${pheno_v3}" \
--mpheno ${m} \
--qcovar "${qcovars}" \
--out "${out}"
done
echo "Done.."Note that in GCTA, the default phenotype index is –mpheno 1. This means that if no –mpheno option is specified, GCTA (fastGWA/fastGWA-GLMM) will run GWAS using the first phenotype in the file.
EXERCISE 10 (Optional): Split GRM Calculation
Goal: Practice splitting GRM computation into multiple parts, running them in parallel, and merging them before creating a sparse GRM.
Show solution
# (Optional) Build GRM in parts, then merge
# Example: split GRM into two parts (these can be run in parallel)
${gcta} --bfile ${geno} --make-grm-part 2 1 --thread-num 8 --out ${output}_grm_partial
${gcta} --bfile ${geno} --make-grm-part 2 2 --thread-num 8 --out ${output}_grm_partial
# Merge GRM parts
cat ${output}_grm_partial.part_2_*.grm.id > ${output}_grm_partial_merged.grm.id
cat ${output}_grm_partial.part_2_*.grm.bin > ${output}_grm_partial_merged.grm.bin
cat ${output}_grm_partial.part_2_*.grm.N.bin > ${output}_grm_partial_merged.grm.N.bin
# Create a sparse GRM
${gcta} --grm ${output}_grm_partial_merged --make-bK-sparse 0.05 --out ${output}_grm_partial_merged.sparse
☕ Coffee break: We will take a 15-minute break here before continuing with the tutorial.
- Introduction to REGENIE
REGENIE is a whole-genome regression method optimised for performing GWAS efficiently in large datasets. It employs a two-step approach: In Step 1, a two-level ridge regression model is fit to a subset of genetic variants to capture polygenic effects. This step generates leave-one-chromosome-out (LOCO) predictions of phenotypes. In Step 2, individual genetic variants are tested for association with the trait, adjusting for the LOCO predictions to account for polygenic effects and potential confounders. The key computational innovation of REGENIE lies in Step 1, where the genotype matrix is divided into large blocks. Each block undergoes first-level ridge regression to generate intermediate predictors, which are then combined in a second-level ridge regression to produce LOCO predictions for the trait. This block-wise parallelisation significantly reduces the computational burden, enabling the method to scale to datasets with hundreds of thousands of samples while preserving accuracy. REGENIE’s efficiency makes it particularly well-suited for large-scale biobank studies, where high computational demands are common.
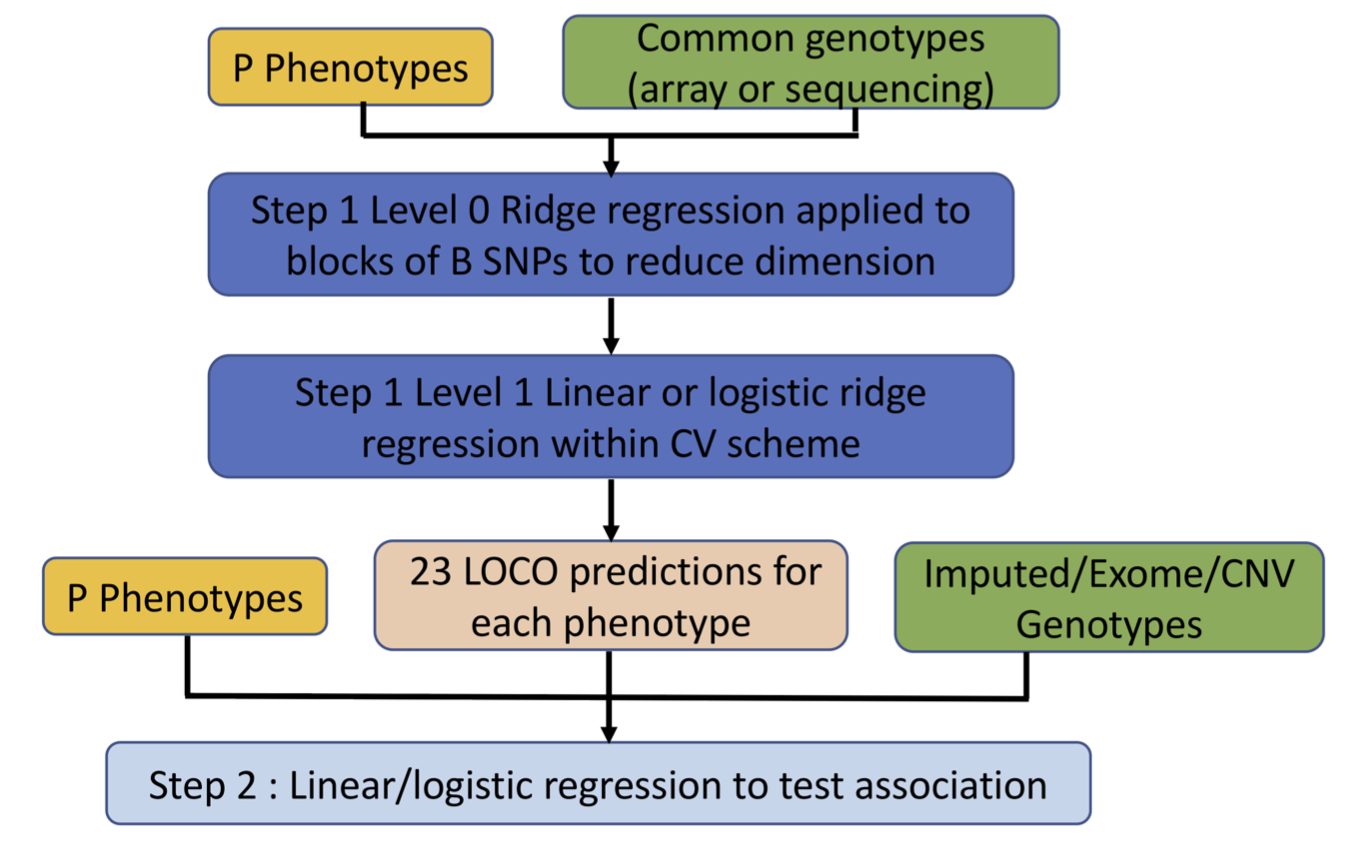
Figure 1. REGENIE Workflow
Mathematical framework of REGENIE
REGENIE conducts whole-genome regression in two computationally efficient steps, making it well-suited for large biobank-scale datasets.
For a quantitative trait, REGENIE begins with the linear mixed model:
\[ y = X\alpha + G \beta + \varepsilon, \tag{1} \]
where \(G\) represents the standardised genotype matrix, \(\alpha\) denotes the fixed effects of covariates, \(\beta\) indicates the effect sizes of genotypes, and \(\varepsilon\) is the random noise, characterised by:
\[ \beta \sim N(0, \sigma_g^2 \mathbf{I}), \quad \varepsilon \sim N(0, \sigma_e^2 \mathbf{I}). \]
For a binary trait, the first stage (Level-0) remains the same, while the second stage (Level-1) employs logistic regression via the logit link. In Step 2, logistic regression is again used for association testing.
Foundations of Step 1 in REGENIE
In Step 1 of REGENIE, consider the following notation:
- \(y \in \mathbb{R}^n\) — phenotype
vector (quantitative or binary, after any required transformation)
- \(X \in \mathbb{R}^{n \times p}\) —
fixed-effect covariates (e.g., age, sex, principal components)
- \(G \in \mathbb{R}^{n \times m}\) —
standardised genotype matrix for \(m\)
variants
- \(\alpha \in \mathbb{R}^p\) —
fixed-effect coefficients
- \(\beta \in \mathbb{R}^m\) —
genome-wide SNP effects
- \(\varepsilon \in \mathbb{R}^n\) — residual errors
-
Simplifying the model
The full model is given by:
\[ y = X \alpha + G \beta + \varepsilon. \]
To remove the contribution of the fixed covariates \(X\), all variables are projected onto the orthogonal complement of the column space of \(X\) using the projection matrix:
\[ P_X = \mathbf{I} - X (X^\top X)^{-1} X^\top. \]
Since \(P_X X = 0\), pre-multiplying the model by \(P_X\) yields:
\[ \widetilde{y} = \widetilde{G} \beta + \widetilde{\varepsilon}, \tag{2} \]
where:
\[ \widetilde{y} = P_X y, \quad \widetilde{G} = P_X G, \quad \widetilde{\varepsilon} = P_X \varepsilon. \]
Equation (2) is therefore the covariate-adjusted model used in Step 1.
-
Level-0 Ridge Regression
The matrix \(\widetilde{G}\) is partitioned into \(B\) non-overlapping blocks:
\[ \widetilde{G} = \big[ \widetilde{G}_1 \; \widetilde{G}_2 \; \cdots \; \widetilde{G}_B \big], \]
where each block \(\widetilde{G}_i \in \mathbb{R}^{n \times m_i}\) contains \(m_i\) variants.
For each block \(\widetilde{G}_i\), ridge regression models are fitted for a set of \(J\) penalty parameters \(\{\lambda_1, \ldots, \lambda_J\}\):
\[ \widehat{\beta}_{i, \lambda_j} = \arg\min_{\beta \in \mathbb{R}^{m_i}} \left\{ \|\widetilde{y} - \widetilde{G}_i \beta\|_2^2 + \lambda_j \|\beta\|_2^2 \right\}. \]
The fitted predictors for block \(i\) and penalty \(\lambda_j\) are:
\[ \widehat{y}_{i, \lambda_j} = \widetilde{G}_i \widehat{\beta}_{i, \lambda_j}. \]
This produces \(J \times B\) predictors in total:
\[ \widehat{Y}^{(0)} = \big[ \widehat{y}_{1,\lambda_1}, \dots, \widehat{y}_{1,\lambda_J}, \widehat{y}_{2,\lambda_1}, \dots, \widehat{y}_{B,\lambda_J} \big]. \]
Since each block can be processed independently, this stage is highly parallelisable, substantially reducing computational cost.
-
Level-1 Ridge Regression (LOCO)
In the second stage, the \(J \times B\) predictors from Level-0 are combined via ridge regression:
\[ \widehat{\gamma} = \arg\min_{\gamma \in \mathbb{R}^{J B}} \left\{ \|\widetilde{y} - \widehat{Y}^{(0)} \gamma\|_2^2 + \lambda^{(1)} \|\gamma\|_2^2 \right\}. \]
Here, \(\lambda^{(1)}\) is selected from a set of shrinkage parameters to minimise the prediction error.
REGENIE employs a Leave-One-Chromosome-Out (LOCO) procedure to avoid proximal contamination:
- For each chromosome \(c\),
predictors derived from variants on chromosome \(c\) are excluded when fitting the Level-1
model.
- The resulting LOCO predictor, \(\widehat{y}^{(1)}_{(-c)}\), is used in
Step 2 when testing variants on chromosome \(c\).
- This generates 22/23 LOCO predictions for a human dataset.
- For each chromosome \(c\),
predictors derived from variants on chromosome \(c\) are excluded when fitting the Level-1
model.
- Running GWAS with REGENIE
EXERCISE 11 (Optional): Running REGENIE Interactively (Quantitative Trait)
dir_data="${dir_user}/snake-cma/examples/data"
dir_cma="${dir_user}/snake-cma/examples/cma-tests"
mkdir -p "${dir_cma}"
geno="${dir_data}/HAPMAP3.qc.genotype"
pheno="${dir_data}/continuous_trait.phen"
covars="${dir_data}/HAPMAP3.covars.cov"
output="${dir_cma}/ex11-regenie-qt"
reg="${dir_user}/snake-cma/examples/software/regenie/regenie_v3.2.1.gz_x86_64_Linux"
plink2="${dir_user}/snake-cma/examples/software/plink2/plink2"
echo "Starting..."# REGENIE Step 1
"${reg}" \
--step 1 \
--bed "${geno}" \
--phenoFile "${pheno}" \
--covarFile "${covars}" \
--bsize 1000 \
--lowmem \
--lowmem-prefix "${output}_step1_cache" \
--out "${output}_step1"# REGENIE Step 2
"${reg}" \
--step 2 \
--bed "${geno}" \
--phenoFile "${pheno}" \
--covarFile "${covars}" \
--pred "${output}_step1_pred.list" \
--bsize 2000 \
--minMAC 20 \
--out "${output}_step2"
echo "Done."EXERCISE 12: Run GWAS with REGENIE for a quantitative trait via a Bash script
Goal: Write and
run a small Bash script that runs REGENIE for a quantitative trait. Make
sure to change the output variable to a unique name before
running.
Show solution
Create the script file:
nano ex12.shPaste the solution below, then save and close:
#!/bin/bash set -euo pipefail dir_user="${1:?ERROR: provide repo root as first argument}" dir_data="${dir_user}/snake-cma/examples/data" dir_cma="${dir_user}/snake-cma/examples/cma-tests" mkdir -p "${dir_cma}" geno="${dir_data}/HAPMAP3.qc.genotype" pheno="${dir_data}/continuous_trait.phen" covars="${dir_data}/HAPMAP3.covars.cov" output="${dir_cma}/ex12-regenie-qt" reg="${dir_user}/snake-cma/examples/software/regenie/regenie_v3.2.1.gz_x86_64_Linux" echo "Starting..." # REGENIE Step 1 "${reg}" \ --step 1 \ --bed "${geno}" \ --phenoFile "${pheno}" \ --covarFile "${covars}" \ --bsize 1000 \ --lowmem \ --lowmem-prefix "${output}_step1_cache" \ --out "${output}_step1" # REGENIE Step 2 "${reg}" \ --step 2 \ --bed "${geno}" \ --phenoFile "${pheno}" \ --covarFile "${covars}" \ --pred "${output}_step1_pred.list" \ --bsize 2000 \ --minMAC 20 \ --out "${output}_step2" echo "Done."Run the script (pass the repository root you saved earlier as dir_user):
bash ex12.sh "${dir_user}"
EXERCISE 13 (Optional): Run GWAS with REGENIE for a quantitative trait using an HPC job script on your cluster
Goal: Write and
run a small job script for your local HPC cluster that runs REGENIE for
a quantitative trait. Make sure to change the output
variable to a unique name before running.
Show solution
A solution for Eddie (the University of Edinburgh HPC cluster) will be provided during the in-person training.EXERCISE 14: Running REGENIE Interactively (Binary Trait)
dir_data="${dir_user}/snake-cma/examples/data"
dir_cma="${dir_user}/snake-cma/examples/cma-tests"
mkdir -p "${dir_cma}"
geno="${dir_data}/HAPMAP3.qc.genotype"
pheno="${dir_data}/binary_trait.phen"
covars="${dir_data}/HAPMAP3.covars.cov"
output="${dir_cma}/ex14-regenie-bt"
reg="${dir_user}/snake-cma/examples/software/regenie/regenie_v3.2.1.gz_x86_64_Linux"
plink2="${dir_user}/snake-cma/examples/software/plink2/plink2"
echo "Starting..."
# REGENIE Step 1
"${reg}" \
--step 1 \
--bed "${geno}" \
--phenoFile "${pheno}" \
--covarFile "${covars}" \
--bsize 1000 \
--lowmem \
--bt \
--lowmem-prefix "${output}_step1_cache" \
--out "${output}_step1"
# REGENIE Step 2
"${reg}" \
--step 2 \
--bed "${geno}" \
--phenoFile "${pheno}" \
--covarFile "${covars}" \
--pred "${output}_step1_pred.list" \
--bsize 2000 \
--minMAC 20 \
--bt \
--firth --approx \
--out "${output}_step2"
echo "Done."EXERCISE 15: Run GWAS with REGENIE for a binary trait via a Bash script
Goal: Write and
run a small Bash script that runs REGENIE for a binary trait. Make sure
to change the output variable to a unique name before
running.
Show solution
Create the script file:
nano ex15.shPaste the solution below, then save and close:
#!/bin/bash set -euo pipefail dir_user="${1:?ERROR: provide repo root as first argument}" dir_data="${dir_user}/snake-cma/examples/data" dir_cma="${dir_user}/snake-cma/examples/cma-tests" mkdir -p "${dir_cma}" geno="${dir_data}/HAPMAP3.qc.genotype" pheno="${dir_data}/binary_trait.phen" covars="${dir_data}/HAPMAP3.covars.cov" output="${dir_cma}/ex15-regenie-bt" reg="${dir_user}/snake-cma/examples/software/regenie/regenie_v3.2.1.gz_x86_64_Linux" echo "Starting..." # REGENIE Step 1 "${reg}" \ --step 1 \ --bed "${geno}" \ --phenoFile "${pheno}" \ --covarFile "${covars}" \ --bsize 1000 \ --lowmem \ --bt \ --lowmem-prefix "${output}_step1_cache" \ --out "${output}_step1" # REGENIE Step 2 "${reg}" \ --step 2 \ --bed "${geno}" \ --phenoFile "${pheno}" \ --covarFile "${covars}" \ --pred "${output}_step1_pred.list" \ --bsize 2000 \ --minMAC 20 \ --bt \ --firth --approx \ --out "${output}_step2" echo "Done."Run the script (pass the repository root you saved earlier as dir_user):
bash ex15.sh "${dir_user}"
EXERCISE 16 (Optional): Run GWAS with REGENIE for a binary trait using an HPC job script on your cluster
Goal: Write and
run a small job script for your local HPC cluster that runs REGENIE for
a binary trait. Make sure to change the output variable to
a unique name before running.
Show solution
A solution for Eddie (the University of Edinburgh HPC cluster) will be provided during the in-person training.
- Introduction to CMA
Corrected Meta-Analysis (CMA) is a divide-and-conquer approach designed for efficient genome-wide association studies on very large datasets. The method splits the data into smaller, more manageable chunks, runs association analyses in parallel using tools such as REGENIE or fastGWA-GLMM, and then combines the results through a novel meta-analysis method that accounts for between-cohort confounders. This workflow reduces computational burden significantly, allows flexible use of high-performance computing resources, and makes large-scale GWAS feasible even with limited per-job memory or runtime constraints.
CMA separates the predictivity stage (estimation of effect sizes) from the discovery stage (identification of loci or generation of p-values). In the predictivity stage, it mitigates the “winner’s curse” effect, providing better-calibrated effect size estimates. In the discovery stage, it achieves equally as good discovery levels as the gold standard GWAS methods such as REGENIE.CMA workflow schematic
| Step | Description |
|---|---|
| Split | Partition cohort into \(M\) sub-cohorts (\(M\) is defined by the user) |
| Analyse | Run GWAS for each sub-cohort in parallel |
| Combine | Merge and meta-analyse results |
\[ \boxed{\quad \text{Data} \quad} \xrightarrow{\mathbf{split}} \boxed{\quad \text{Sub-cohort 1},\ \text{Sub-cohort 2},\ \ldots \quad} \xrightarrow{\mathbf{GWAS\ (in\ parallel)}} \boxed{\quad \widehat{\beta}_1,\ \widehat{\beta}_2,\ \ldots \quad} \xrightarrow{\mathbf{corrected\ meta\text{-}analysis}} \boxed{\quad \widehat{\beta}_{\text{final}} \quad} \]
Quantifying Relationships Between Sub-cohorts in CMA
When combining results from multiple sub-cohorts into a single estimate, it is important to account for the degree of overlap in information between sub-cohorts, which can arise from the presence of related individuals distributed across different sub-cohorts.. Overlap affects how similar the separate results are to one another, and failing to adjust for it can bias the combined result.
CMA incorporates this overlap adjustment through the following relationship:
\[ \mathrm{Corr}(\widehat{\beta}_i, \widehat{\beta}_j) = \gamma_{12} \, \mathrm{Corr}(z_i, z_j), \]
where:
- \(z_i\) and \(z_j\) are the z-scores from the GWAS results of cohorts \(i\) and \(j\), respectively,
- \(\gamma_{12}\) is defined as: \[ \gamma_{12} = \begin{cases} -1, & \text{if } \widehat{\beta}_i \cdot \widehat{\beta}_j < 0, \\ \;\;1, & \text{otherwise}. \end{cases} \]
CMA Predictivity
By modelling and adjusting for the correlation structure arising from cohort overlap, CMA produces effect size estimates that are better calibrated and less biased compared to standard GWAS. This yields improved predictive performance when the resulting polygenic scores are applied to independent datasets.
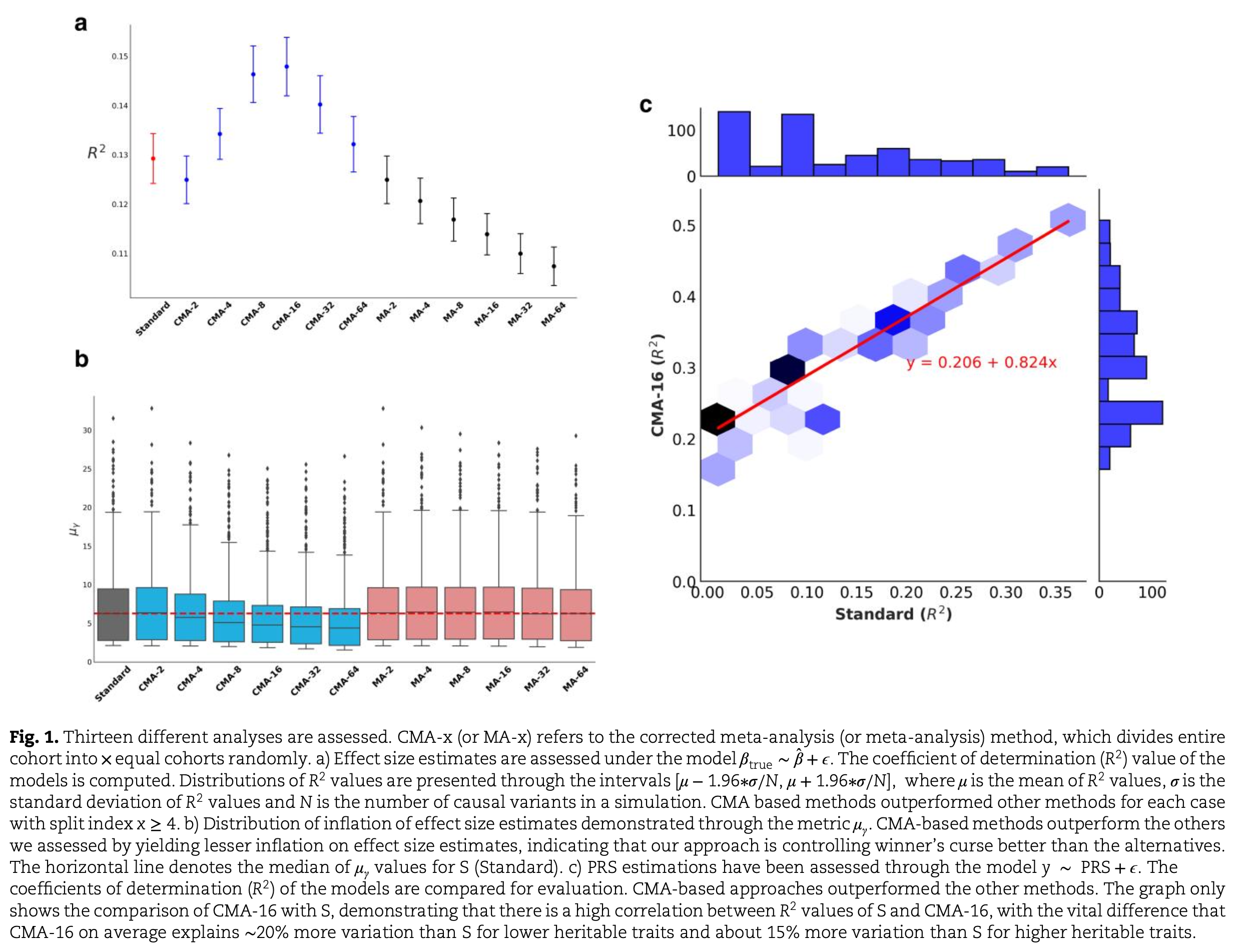
CMA Discovery
CMA achieves lower false discovery rates than standard GWAS methods, although this comes at the expense of some statistical power. For those prioritising higher power, CMA includes an inflation-based summary statistics option that enables discovery levels comparable to standard GWAS approaches. This is made possible by CMA’s decoupled framework for prediction and discovery.
We applied CMA to 12 traits from the UK Biobank using the inflation mode. CMA maintained discovery power equivalent to that of REGENIE in terms of genome-wide significant loci.
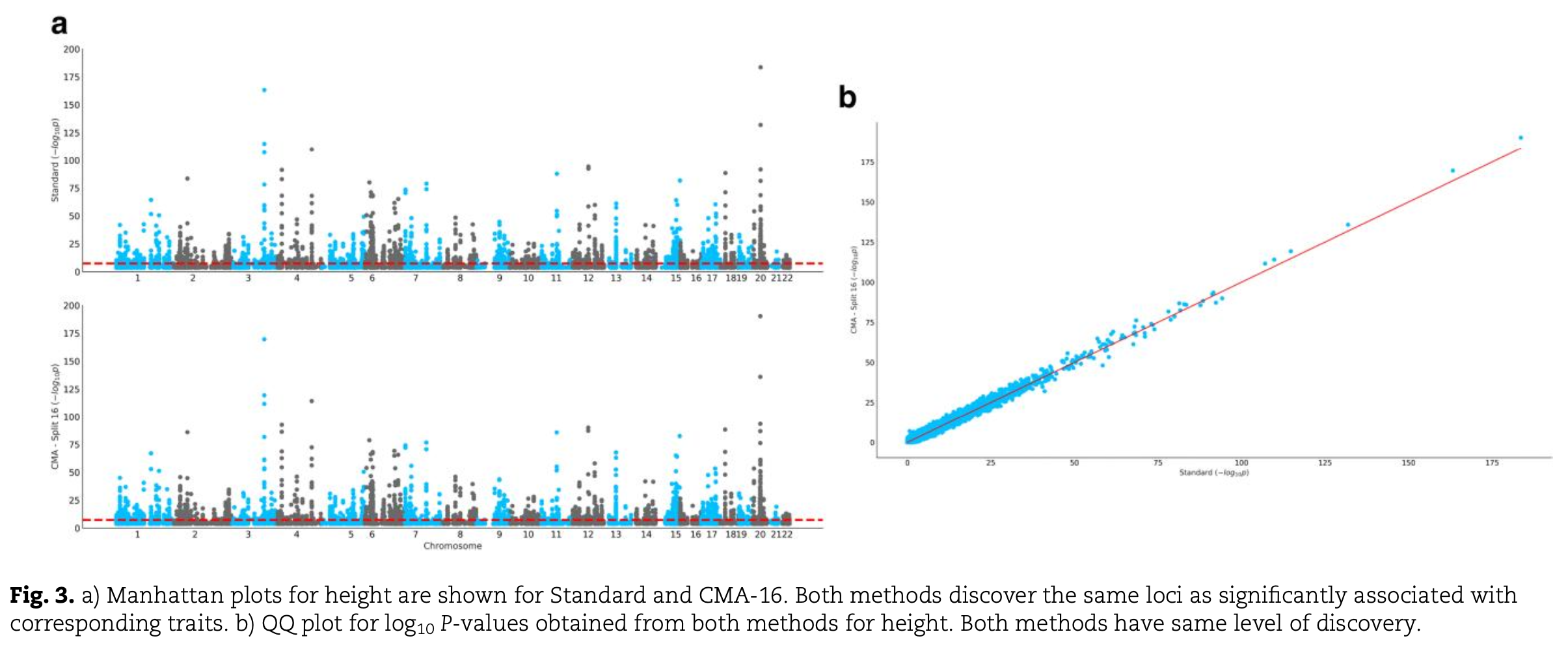
A Manhattan plot is a scatter plot commonly used in GWAS to display –log₁₀ p-values of genetic variants across the genome, with chromosomes arranged sequentially along the x-axis. It is used to visualise and identify genomic regions with strong association signals. A Q–Q plot compares the observed distribution of test statistics (such as p-values) with the expected distribution under the null hypothesis. In the figure above, it is used to compare the distributions of GWAS summary results obtained from CMA and REGENIE.
Computational Benchmark of CMA
Extensive benchmarking demonstrates that CMA offers substantial computational advantages over both REGENIE and fastGWA-GLMM. It enables GWAS to be performed on large-scale datasets using only moderate computing resources if required; for example, CMA can run efficiently on datasets containing 5 million individuals.
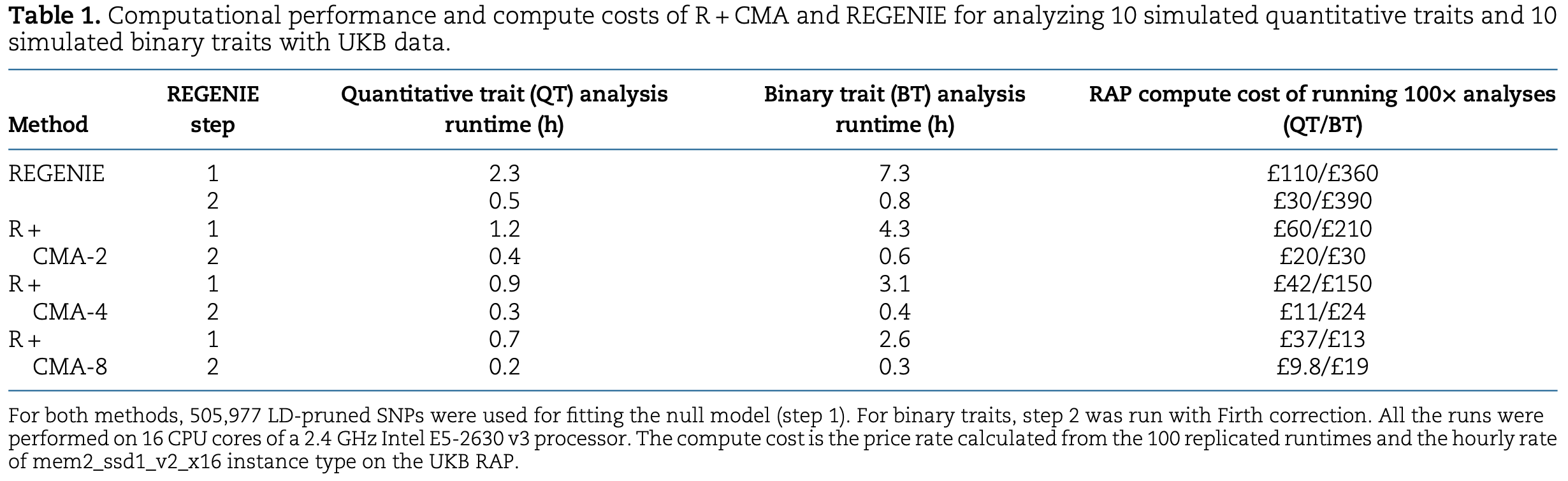
R + CMA (or RCMA in short) indicates that CMA is used in conjunction with REGENIE to perform GWAS on sub-cohorts.
CMA as a Multi-Purpose Tool
CMA is a versatile framework for large-scale genetic association studies, offering both GWAS execution and meta-analysis within the same framework. For GWAS, CMA supports running REGENIE (RCMA) and fastGWA-GLMM (FCMA) directly with a split of 1 (i.e. \(M=1\)), producing results identical to running each method natively. When a split index greater than 1 is specified, CMA automatically partitions the dataset, runs GWAS on each subset in parallel, and then combines the results, enabling efficient analysis of datasets without loosing any statistical discovery. In addition, CMA implements a unique meta-analysis method that corrects for related individuals across sub-cohorts, currently the only approach offering this capability.
Key capabilities of CMA:
- GWAS modes:
- REGENIE (RCMA with split = 1)
- fastGWA-GLMM (FCMA with split = 1)
- RCMA/FCMA with split > 1
- REGENIE (RCMA with split = 1)
- Meta-analysis mode (Not covered in this tutorial):
- CMA that accounts for relatedness between sub-cohorts
- Running GWAS with CMA
EXERCISE 17: Run GWAS with CMA via a Bash script
Goal: Write and run a small job script for CMA, using either R + CMA (RCMA) or F + CMA (FCMA), which splits the cohort into 2 sub-cohorts for a binary trait.
Show solution
The downloaded repository already includes the necessary Bash
scripts, with RCMA and FCMA options providing the solution to this
exercise. The script rcma.qt.sh can also be tested on a
quantitative trait, although this run takes considerably longer to
complete.
# Option A: R+CMA — REGENIE (Quantitative)
bash ./snake-cma/examples/rcma.qt.sh
# Option B: R+CMA — REGENIE (Binary)
bash ./snake-cma/examples/rcma.bt.sh
# Option C: F+CMA — fastGWA-GLMM (Binary)
bash ./snake-cma/examples/fcma.bt.shEXERCISE 18: Run REGENIE with CMA
Goal: Run R + CMA (RCMA) with a single split (i.e. \(M=1\)) to execute REGENIE under the CMA framework.
Show solution
dir_data=${dir_user}/snake-cma/examples/data
dir_cma=${dir_user}/snake-cma/examples/cma-tests
mkdir -p ${dir_cma}
geno=${dir_data}/HAPMAP3.qc.genotype
pheno=${dir_data}/binary_trait.phen
covars=${dir_data}/HAPMAP3.covars.cov
output=${dir_cma}/ex18-rcma-bt-split1
reg=${dir_user}/snake-cma/examples/software/regenie/regenie_v3.2.1.gz_x86_64_Linux
plink2=${dir_user}/snake-cma/examples/software/plink2/plink2
cma=${dir_user}/snake-cma/src/snake_cma.py
echo Starting..
python ${cma} \
--bed ${geno} \
--bt \
--split 1 \
--out ${output} \
--phenoFile ${pheno} \
--covarFile ${covars} \
--regenie ${reg} \
--plink ${plink2}
echo Done..EXERCISE 19: Run fastGWA-GLMM with CMA
Goal: Run F + CMA (FCMA) with a single split (i.e. \(M=1\)) to execute fastGWA-GLMM under the CMA framework.
Show solution
dir_data=${dir_user}/snake-cma/examples/data
dir_cma=${dir_user}/snake-cma/examples/cma-tests
mkdir -p ${dir_cma}
geno=${dir_data}/HAPMAP3.qc.genotype
pheno=${dir_data}/binary_trait.phen
covars=${dir_data}/HAPMAP3.covars.qcovar
output=${dir_cma}/ex19-fcma-bt-split1
reg=${dir_user}/snake-cma/examples/software/regenie/regenie_v3.2.1.gz_x86_64_Linux
plink2=${dir_user}/snake-cma/examples/software/plink2/plink2
cma=${dir_user}/snake-cma/src/snake_cma.py
echo Starting..
# Build sparse GRM only if it doesn't exist
if [ ! -f "${grm}.grm.sp" ]; then
${gcta} --bfile ${geno} \
--make-grm \
--sparse-cutoff 0.05 \
--out ${grm}
fi
python ${cma} \
--bt \
--bed ${geno} \
--sp-grm ${grm} \
--split 1 \
--out ${output} \
--phenoFile ${pheno} \
--covarFile ${covars} \
--gcta ${gcta} \
--plink ${plink2} \
--joint_covar
echo Done..
- (Optional) Running Meta-Analysis with CMA
EXERCISE 20: Run Meta-Analysis with CMA via a Bash script
Goal: Combine GWAS results from fastGWA-GLMM or REGENIE using the CMA meta-analysis approach.
Show solution
# Option D: CMA Meta-analysis — Quantitative
bash ./snake-cma/examples/cma.qt.sh
# Option E: CMA Meta-analysis — Binary
bash ./snake-cma/examples/cma.bt.sh💬 Questions / Comments / Feedback
Thank you for joining this training session on Scalable Methods for Large-Scale Genetic Association Studies.
In this session, we covered three major GWAS tools REGENIE, fastGWA/fastGWA-GLMM, and CMA, explored their core concepts, and showed how to run them through hands-on examples with sample datasets.This concludes our tutorial. Now is the time for any remaining questions or comments before we wrap up.
If you have questions in the future or need additional clarification, please don’t hesitate to reach out:
İsmail Özkaraca: ozkaraca AT duck DOT com
References
[1] (REGENIE) Mbatchou, J., Barnard, L., Backman, J. et al. Computationally efficient whole-genome regression for quantitative and binary traits. Nat Genet 53, 1097–1103 (2021). https://doi.org/10.1038/s41588-021-00870-7
[2] (fastGWA) Jiang, L., Zheng, Z., Qi, T. et al. A resource-efficient tool for mixed model association analysis of large-scale data. Nat Genet 51, 1749–1755 (2019). https://doi.org/10.1038/s41588-019-0530-8
[3] (fastGWA-GLMM) Jiang, L., Zheng, Z., Fang, H. et al. A generalized linear mixed model association tool for biobank-scale data. Nat Genet 53, 1616–1621 (2021). https://doi.org/10.1038/s41588-021-00954-4
[4] (GCTA) Yang, Jian, et al. “GCTA: a tool for genome-wide complex trait analysis.” The American Journal of Human Genetics 88.1 (2011): 76-82. https://doi.org/10.1016/j.ajhg.2010.11.011
[5] (CMA) Mustafa İsmail Özkaraca, Mulya Agung, Pau Navarro, Albert Tenesa, Divide and conquer approach for genome-wide association studies, Genetics, Volume 229, Issue 4, April 2025, iyaf019, https://doi.org/10.1093/genetics/iyaf019
[6] Listgarten, J., Lippert, C., Kadie, C. et al. Improved linear mixed models for genome-wide association studies. Nat Methods 9, 525–526 (2012). https://doi.org/10.1038/nmeth.2037